![[title]](images/title.gif)
|
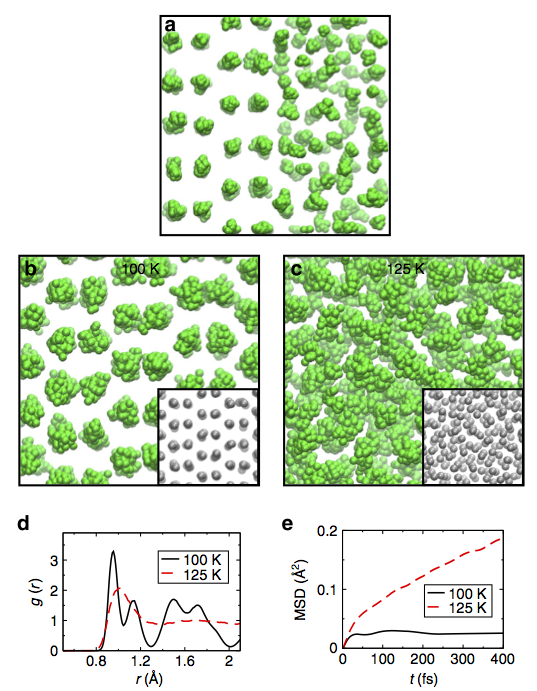
We study the structure and dynamical properties of materials using a form of quantum mechanics known as Density Functional Theory (DFT). The group are major developers of the general-purpose materials simulation package CASTEP which is used world-wide. As such, the group is active in both developing and coding new algorithms, as well as applications of CASTEP. Examples of recent algorithmic developments include a new 'auxilliary functional' approach to solving the key equations of DFT which provide better robustness and speed than conventional approaches (Hasnip and Probert). We have also pioneered new approaches to structure prediction using a novel periodic genetic algorithm (Abraham and Probert). Examples of recent applications include combining the power of DFT with Path Integral Molecular Dynamics (PIMD) to study the properties of hydrogen. Recent studies include the high-pressure phase diagram of hydrogen and the study of hydrogen diffusion on a metal surface. The picture shows the use of two-phase coexistence with PIMD to study the high pressure melting of hydrogen. These studies were combined with theoretical colleagues from Cambridge, UCL and the University of Beijing. Other recent applications include studies of defects in various 2D materials, such as graphene nanoribbons or MoS2 sheets, and combining the power of DFT with experimental data to elucidate the structure of defects. Again, these studies were done in collaboration with experimental colleagues in York, Saudi Arabia and China. The following is a brief description of some of our current projects: |
|
Density Functional Theory and CASTEP
Phil Hasnip and Matt Probert
Density Functional Theory (DFT) is a Nobel-prize winning reformulation of quantum mechanics in terms of electron density rather than wavefunctions. This results in a huge reduction in complexity and enables efficient numerical implementation, so that computer codes (e.g. CASTEP) can now calculate from first principles (i.e. ab initio) the properties of many materials. There have been many books and review articles written on the fundamental theory.
CASTEP, someone once said, is a "fluffy Fortran mega-monster written by a collection of strange men", which, whilst this may be true, is not the most helpful description. It would be more accurate to say that CASTEP is a complex and powerful computer program that uses DFT to simulate the electronic, optical and structural properties of solids, interfaces, and surfaces for a wide range of materials classes including ceramics, semiconductors, and metals. The quantum mechanical accuracy of this approach allows many material and chemical properties to be predicted with no experimental input, providing insight into physical phenomena even under extreme conditions that cannot be reproduced in the laboratory. The code has been developed by a number of academics over the years, known collectively as the CASTEP Developer Group (CDG) including many contributions from this group. See below for more details.
|
Phil's research focuses on developing and optimising algorithms and computational methods for CASTEP. Recent applications include simulations of half-metallic Heusler alloys for use in future spintronic devices, and predicted electron-energy loss spectra (EELS) from graphene nanoribbons. The main goal of this research is to make these calculations both robust and efficient regardless of the material or chemical being simulated, and on all classes of computing hardware, from few-core PCs to massively-parallel High Performance Computers. This requires developments in a wide variety of fields, from non-linear optimisation and eigenvalue solvers to parallel programming, all underpinned by good software engineering and design principles to ensure the program is as bug-free, portable and maintainable as possible. The animated image shows an electron cloud condensing onto sodium phenoxide over successive iterations of a CASTEP ground state calculation. Sodium phenoxide has covalent C-C and C-H bonds, a delocalised metallic-like pi-cloud, a polar C-O bond and an ionic O- Na+ bond, all of which emerge spontaneously during the CASTEP calculation. |

|
Path Integral Molecular Dynamics and Quantum Diffusion on Surfaces
Aaron Hopkinson, Joly Aarons and Matt Probert
|
The properties of materials at surfaces will differ from those of the bulk, due to the change in environment at the interface. While we may learn a lot about a material by studying its bulk properties, the study of surfaces is necessary if we wish to fully understand the world around us, since real materials do form surfaces. Furthermore, knowledge about surface properties is essential for the study of processes involving friction and many catalytic reactions. Recent work within the group has been to study the diffusion of hydrogen on metal surfaces using a variety of approaches including density functional theory and path integral molecular dynamics to gain an understanding of atomic scale temperature dependent friction. Though the nuclei of light atoms are often assumed to behave as classical particles, this is in general untrue. Indeed, hydrogen exhibits a strong degree of nuclear quantum delocalisation even at room temperature. This can be captured by the technique of path integral molecular dynamics (PIMD), where the system under study is represented at several difference points in imaginary time which represent different possible positions in the quantum spread; quantum ensemble averages of system properties may then taken. We also exploit recent advances to incorporate real time quantum dynamics using such techniques as partially adiabatic centroid PIMD. |
|
Structure Prediction and Global Optimisation
Ed Higgins and Matt Probert
|
The idea of global structure prediction, i.e. finding the ground state structure of a material based simply on its constituent atoms, has been around for a while now. The periodic genetic algorithm approach of Abraham and Probert, and the ab initio random structure search of Pickard and Needs, have been at the forefront of developing new techniques, and both approaches have used CASTEP. However there are many useful materials that, while not being in their ground state, are both thermodynamically stable and incredibly useful. This is particularly evident in the wide variety of carbon allotropes, both used in industry and actively researched across the world. This project is developing and implementing a multi-objective genetic algorithm for crystal structure prediction, based on work previously done in the group for the CASTEP code. This will allow structural optimisation of many physical properties, in addition to minimising the structure's energy. In particular, the hardness of Carbon Nitride allotropes is being investigated. |
|
Shock Waves
Jacob Wilkins and Matt Probert
|
Shock waves are extreme events in nature that have a wide range of applications in areas such as geophysics, astrophysics and in industry. Our research is to use atomistic computer simulations to probe high-temperature and high-pressure properties of condensed matter that occur when it is subjected to a "shock". It is our goal to achieve a greater understanding of condensed matter when it is in these extreme states. |

|
This project is based on exploration of various approaches to shock-wave simulations, such as Non-Equilibrium MD (NEMD), with the hope of making these simulations cheaper, while maintaining accuracy, and allowing more detailed calculations. The project aims to extend recently developed techniques such as neural network potentials and Gaussian approximation potentials to high-pressure, high-energy systems and compare with other approaches already in use in the area such as direct NEMD and the recently developed Hugoniostat. This should make it possible to achieve DFT accuracy in shock simulations without paying the high computational cost. Possible applications of this range from: new methods for accurate large-scale molecular dynamics, to geophysics and high-energy physics, where many phenomena occur very quickly or on large spatial scales making normal DFT calculations infeasible.
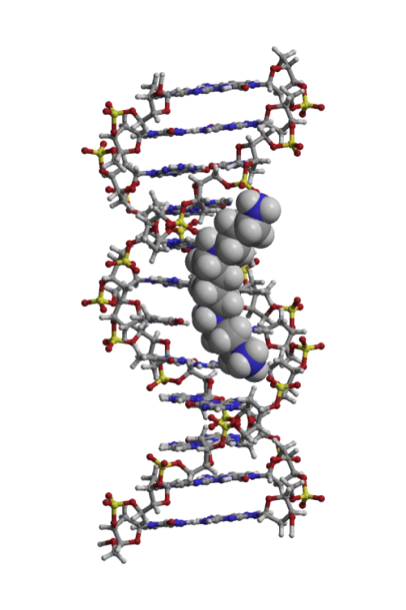
Biological Physics
Jack Shepherd and Matt Probert
|
The modus operandi of DNA is well known, but the core physics behind these interactions and processes are little understood. A general understanding of the fundamental science at work would allow better bionanotechnology to be designed, and would open the door to a new era of therapeutic applications of biological physics. This has historically been limited by a disconnect in how the subcellular world has been explored. Either experiments were undertaken, which were not sophisticated enough to give fine detail, or simulations of biological systems were undertaken, but contemporary computing limited these to systems so small as to be all but irrelevant biologically. The scientific community was traditionally split between detailed but tiny simulation and broad-scale but low resolution experiment. However, it is now possible to perform an all-atom simulation on tens or hundreds of thousands of atoms, and even a full quantum mechanical calculation may be done on systems of thousands of atoms. New coarse-grained simulation software produced recently allows thousands of base pairs of DNA to be simulated and its broad features extracted. Similarly, the limits of what may be seen in a microscope have receded, and using bespoke microscopes and cutting-edge superresolution techniques, individual fluorescent molecules may be pinpointed. Newly designed optical and magnetic tweezers are being constructed which will permit very fine manipulation of individual DNA strands. This project combines multiple approaches so that a more full picture of DNA dynamics may be built up than has been possible previously. Beginning with investigation of the structure of classic double-stranded double helix DNA, it is anticipated that this investigation will progress on to working with molecular machines to understand the forces which drive them and, in turn, maintain life. |
|
CASTEP uses the total energy method, with dual-grid plane-wave basis sets and ultrasoft pseudopotentials. The electronic energy may be minimised using the conjugate gradient technique with either density mixing, ensemble DFT or an all-bands formulation. Once the ground state electronic energy is known, it may then be used to calculate many properties of interest, including electronic, optical and phonon properties. It can also be used to calculate the forces on each atom and the stresses on the unit simulation cell, which can then be used to relax the ionic configuration to the minimum energy and stress configuration using various geometry optimization techniques such as BFGS or damped MD. The main thrust of this research group though is dynamical phenomena, and so our primary interest is in developing new MD simulation techniques and applying them to the study of dynamical processes.
|
CASTEP is a highly modular Fortran90 code, written using object oriented techniques. It has been designed to be highly portable and has been used for research on many different platforms, from Linux/Mac/Windows PCs up to the largest national supercomputers in the world. It has been optimised for single processor performance, and for multi-core performance using OpenMP, and also to scale to massive core counts with parallel efficiency using MPI. Matt and Phil are two of the key members of the CASTEP Developers Group (CDG). |
|
|
Matt has been the Chair of the UKCP consortium since 2006. This is a leading High End Computing consortium, sponsored by EPSRC, and which has had CASTEP as its flagship code for many years. Consequently, CASTEP is freely available to all UK academics through a special arrangement between UKCP and Accelrys Inc who also distribute the code commercial with a graphical frontend for MS Windows known as Materials Studio. |
|